- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2008
- Structural biology
- Atomic structure of a molecular switch for blood coagulation and fibrinolysis
Atomic structure of a molecular switch for blood coagulation and fibrinolysis
After blood-vessel injury, hemostasis induces fibrin clot formation to prevent blood loss and trigger vessel repair. This clot must be removed after tissue repair to restore blood flow. These two processes are tightly regulated by the coagulation and fibrinolytic cascades because imbalance may lead to thrombosis, heart attack and stroke, or to bleeding as in hemophilia [1]. Hemostasis is modulated by thrombin-activatable fibrinolysis inhibitor (TAFI). It attenuates fibrinolysis by removing surface-exposed C-terminal lysine residues from the fibrin clot. TAFI is the zymogen of a B-type zinc-dependent metallocarboxypeptidase of the funnelin tribe of proteases [2,3] and it is the only funnelin found in human plasma. During coagulation, TAFI is processed by thrombin/ thrombomodulin to TAFIa through removal of its pro-domain. In contrast to pancreatic funnelins, both TAFI and TAFIa uniquely display carboxypeptidase activity against larger substrates. However, while TAFIa has a half-life of less than ten minutes, TAFI is stable in circulation [4,5].
 |
|
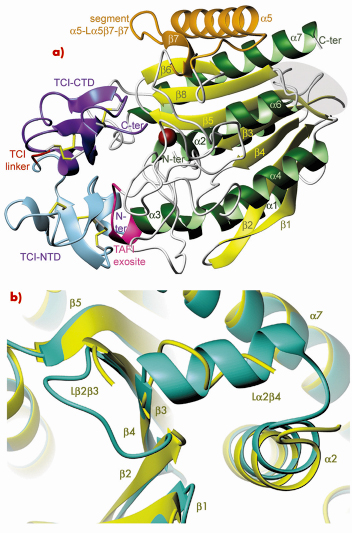
Fig. 76: Structure of the TAFIa/TCI complex. a) Richardson plot of the complex showing TAFIa in standard orientation. The region of the proposed fibrinolysis switch is shown over gray background. TCI is coloured as follows: N-terminal domain (light blue), linker (red), C-terminal domain (purple). b) Close-up view of (a) after a vertical rotation of ~90º showing the proposed fibrinolysis switch region of TAFIa (yellow) superimposed with the equivalent region of human pancreatic CPB1, which is taken as a highly-stable reference (green). |
We obtained a stable form of TAFIa in vitro employing purified bovine TAFI, which was activated in the presence of a proteinaceous inhibitor, TCI, from the tick. This produced a stable enzyme-inhibitor complex. We were able to solve the crystal structure to high resolution using synchrotron radiation at beamline ID23-2. These studies revealed that TAFIa conforms to the α/ß-hydrolase fold of funnelins and that it displays two unique flexible loops on the molecular surface, which account for structural instability and susceptibility to proteolysis (Figure 76). In addition, point mutations reported to enhance protein stability in vivo are mainly located in the first loop and in another surface region, which is a potential heparin-binding site. The protein inhibitor contacts both the TAFIa active site and an exosite, thus contributing to high inhibitory efficiency.
Principal publication and authors
L. Sanglas (a), Z. Valnickova (b), J.L. Arolas (c), I. Pallarés (a), T. Guevara (c), M. Solà (a), T. Kristensen (b), J.J. Enghild (b), F.X. Aviles (a) and F.X. Gomis-Rüth (c). Mol. Cell 31, 598 (2008).
(a) Institut de Biotecnologia i de Biomedicina and Departament de Bioquímica i Biologia Molecular, Universitat Autònoma de Barcelona, Bellaterra (Spain)
(b) Center for Insoluble Protein Structures at the Department of Molecular Biology, University of Århus (Denmark)
(c) Department of Structural Biology at the Molecular Biology Institute of Barcelona, CSIC (Spain).
References
[1] M.B. Boffa, M.E. Nesheim and M.L. Koschinsky, Curr. Drug Targets Cardiovasc. Haematol. Disord. 1, 59 (2001).
[2] J.L. Arolas, J. Vendrell, F.X. Avilés and L.D. Fricker, Curr. Pharm. Des. 13, 349 (2007).
[3] F.X. Gomis-Rüth, Crit. Rev. Biochem. Mol. Biol. 43, 319 (2008).
[4] M.B. Boffa, W. Wang, L. Bajzar and M.E. Nesheim, J. Biol. Chem. 273, 2127 (1998).
[5] Z. Valnickova, I.B. Thogersen, J. Potempa and J.J. Enghild, J. Biol. Chem. 282, 3066 (2007).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.