- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2007
- Surface and Interface Science
- Direct detection of the structure-activity correlation in heterogeneous catalysis
Direct detection of the structure-activity correlation in heterogeneous catalysis
A major challenge in (heterogeneous) catalysis research is the identification of the catalytically-active phase under practical reaction conditions. Frequently, the active phase is only stable under reaction conditions and may even change when the reactant mixture or the reaction temperature is varied. All these complications call for in situ characterisation of the catalytic system. To have full access to the atomic-scale structure of the catalyst one has to study model catalysts with low structural complexity such as single crystalline films. One has to pay for this simplification by the introduction of the so-called materials gap that has to be bridged in order to transfer atomic-scale information of the model catalyst to the practical catalyst consisting of metal nanoparticles dispersed on a support material.
With the CO oxidation reaction over RuO2(110)/Ru(0001), we are in the fortunate situation that both the pressure and the materials gaps have been successfully bridged [1]. With the unique high-pressure chamber of beamline ID03 [2], we were able to follow the surface structure of the Ru(0001)-based model catalyst by SXRD and the reaction rate of the product formation (CO2) by mass spectrometry simultaneously during the catalysed CO oxidation reaction over RuO2(110)/Ru(0001) under practical reaction conditions. In these experiments the high-pressure cell was used as a batch reactor.
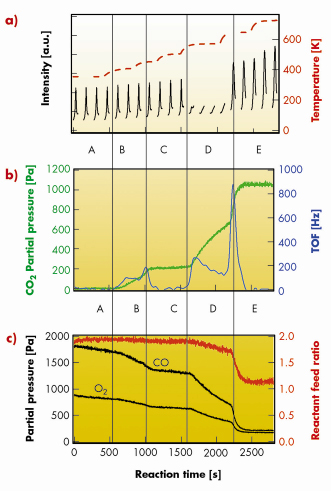
In Figure 90 we present a specific set of experiments aimed at elucidating the structure-activity relationship for the CO oxidation over the RuO2(110)/Ru(0001) model catalyst. The 1.6 nm thick RuO2(110) film on Ru(0001) was produced prior to the reaction experiments by exposing the Ru(0001) surface to 1000 Pa of O2 at 640 K. The sample was then cooled to 350 K and 1800 Pa of CO was admitted to the reaction cell. Subsequently the reaction temperature was increased stepwise up to 720 K over a time period of 2800 s (Figure 90a, brown trace). At temperatures up to 500 K, the oxide film does not change as indicated by the repetitive h-scans (Figure 90a, black traces); the maximum intensity is found at h = 0.73 characteristic for the RuO2(110) oxide surface [3]. For T = 350 K, almost no CO2 is formed (Figure 90b, green trace) as indicated by a small value (< 5 Hz) of the turn over frequency (TOF). Above 400 K the CO2 production is accelerated, revealing a constant TOF of 95 Hz (at 400 K) which increases to 190 Hz by raising the temperature to 450 K. Both CO and O2 are consumed during the CO oxidation, but the stoichiometry of the reaction feed is nearly preserved during the first 2200 s as indicated in Figure 90c (red trace). In region C, the CO2 production stops, although the temperature is increased to 500 K. Presumably, a deactivation of the catalyst occurs whose origin is not understood. At the end of region C (reaction time about 1600 s), the oxide film disappeared. Obviously, below 500 K the catalyst was stabilised in a metastable phase (oxide) in a reducing reactant mixture. Upon reduction of RuO2(110) the activity increased substantially: TOF = 260 Hz. When the temperature was further increased to 640 K, the oxide reappeared (with higher diffraction intensity at h = 0.73). From corresponding l-scans we derive a thickness of the RuO2(110) film of 2.3 nm. The oxidation process of Ru(0001) is accompanied by a sudden rise of the TOF value to 850 Hz. With this increase of the TOF the stoichiometry of the reactant feed drops significantly from 1.9 to 1.15, i.e. the reaction mixture is now much more oxidising than during the first 2000 s (Figure 90c, red trace). This change in the stoichiometry of the reactant feed may cause the rapid TOF decline to very small values <1 Hz. Practically the CO oxidation reaction stops although both CO (210 Pa) and O2 (170 Pa) are still present in the gas mixture. A further increase of the temperature to 720 K did not restore the activity of the RuO2(110) catalyst. The catalyst is deactivated either structurally or by the product CO2 in the gas atmosphere.
 |
|
Fig. 90: The reaction cell is run as a batch reactor and both structural (a) and reactive properties (b, c) are monitored in situ by SXRD and mass spectrometry, respectively, during the CO oxidation reaction over RuO2(110)/Ru(0001). Details are given in the text. |
These particular experiments already demonstrate a complex structure-activity relationship even in the seemingly simple CO oxidation over RuO2(110)/Ru(0001). The activity of the catalyst increases substantially in a transient way whenever the catalyst undergoes structural changes either by reducing RuO2(110) or by forming RuO2(110). At a temperature of 400 K, the (metastable) oxide catalyst works stably with a medium high activity. Deactivation of the catalyst appears in the temperature region of 450 K to 500 K. Complete deactivation is observed above 650 K and under strongly oxidising reaction conditions (P(CO)/P(O2) = 1.15).
References
[1] H. Over and M. Muhler, Prog. Surf. Sci. 72, 3 (2003).
[2] P. Bernhard et al., Rev. Sci. Instrumen. 70, 1478 (1999).
[3] Y.B. He, M. Knapp, E. Lundgren, H. Over, J. Phys. Chem. B 109, 21825 (2005).
Authors
H. Over (a), O. Balmes (b), N. Bovet (c), E. Lundgren (c).
(a) Physikalisch-Chemisches Institut, Justus Liebig University Giessen (Germany)
(b) ESRF
(c) Dept. of Synchrotron Radiation Research, University of Lund (Sweden)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.