- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2007
- Structural Biology
- Redox-mediated substrate recognition by Sdp1 defines a new group of tyrosine phosphatases
Redox-mediated substrate recognition by Sdp1 defines a new group of tyrosine phosphatases
Eukaryotic organisms have evolved sophisticated cellular machinery that allows individual cells to respond to challenges, or stresses, coming from their environment. These stress-response systems work through a network of signal transduction pathways, which alert the cell to the presence of stress factors, and coordinate an appropriate response. Stress-response pathways can be triggered by a diverse range of stimuli that are potentially harmful to the cell, including UV radiation, oxidative and osmotic stress, heat shock and exposure to certain genotoxic substances. The physiological outcome can vary from a transient up-regulation of mechanisms to combat the immediate challenge, to growth arrest and programmed cell death – which prevents damaged cells from reproducing further. The fine control of this balance between cell survival and cell death has potential implications in the treatment of human cancers, as the cellular response to chemotherapeutic agents or radiotherapy may be partly orchestrated by stress-response mechanisms. This, together with studies that have linked components of mammalian stress-response pathways to tumorigenesis, has sparked interest in stress-response mechanisms in recent years.
At the heart of the signalling networks that sense cellular exposure to environmental stress is a highly conserved module known as the mitogen-activated protein kinase (MAPK) cascade. The components of this cascade are enzymes that transduce and amplify signals using a phosphorylation mechanism involving the sequential activation of each protein in the pathway by the addition of phosphate. The magnitude and duration of signalling through the cascade depends on the nature of the incoming signal and the type of cell, and is a key factor in determining the cellular response. The cell has to be able to switch-off or regulate signalling through the cascade, once the initial sensing and signalling event has passed. A family of proteins known as the MAPK phosphatases (MKPs) carry out this function by dephosphorylating and thus inactivating specific MAPKs.
MKPs are cysteine-based phosphatases (CBPs) and possess a cysteine sulphydryl group that plays a central role in catalysis. Sulphydryl groups are sensitive to oxidation, which poses a conundrum for MKPs when the activating stimulus for their target MAPK involves reactive oxygen species (ROS). Under cellular stress induced by oxidative conditions the activity of CBPs would be expected to be temporarily lost; potentially leading to sustained or uncontrolled stress-induced MAPK signalling. We hypothesised that MKPs acting on stress-response pathways stimulated by ROS would present novel solutions to this apparent paradox.
To test this theory we targeted an MKP from yeast called Sdp1, which acts on a MAPK pathway activated by oxidative stress. Surprisingly, we found that this phosphatase exhibits increased activity under oxidative conditions. In stark contrast to other CBPs, the Michaelis constant of Sdp1 increased markedly in the absence of reducing agents, indicating that oxidation actually boosts the activity. Mutational and mass spectrometric analysis pinpointed a potential disulfide-bridge between two cysteines residues (Cys47 and Cys142) as the principle mediator of this effect.
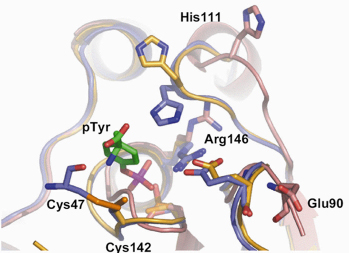
To unravel the molecular basis of the mechanism, we crystallised Sdp1 in oxidising and reducing conditions and in the presence of a phosphotyrosine substrate. Data collected at the ESRF revealed that the proposed disulphide-bridge between Cys47 and Cys142 did occur under oxidising conditions (Figure 76). Moreover, this disulfide and a near-by histidine residue (His111) form a deep cleft that controls access to the active site of the enzyme, thus providing substrate selectivity. In oxidised crystals both the Cys47-Cys142 disulphide and His111 were in direct contact with the phosphotyrosine substrate. This interaction suggests that the boost in activity under oxidising conditions results from the formation of the intramolecular disulphide-bridge, which increases the enzyme’s substrate affinity. The disulphide was not observed in crystals grown in reducing conditions indicating that Sdp1 switches from a high-activity to a low-activity form, when the cell returns to normoxic conditions.
 |
|
Fig. 76: The active site of Sdp1 in low and high activity states. The key disulphide between cysteine 47 and cysteine 142 that mediates the oxidative activation mechanism is shown in gold. |
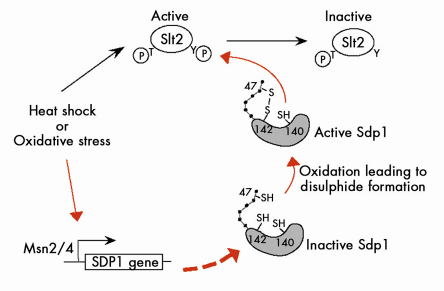
This redox-mediated activation mechanism was unprecedented for a protein tyrosine phosphatase and immediately raised the question as to whether the mechanism was unique to Sdp1. Using sequence homology searches we identified a family of proteins in yeast and filamentous fungi taxa with similar sequence motifs around the substrate access cleft. We have suggested the term “WH-phosphatases” to classify this previously unrecognised group and have shown that one of the paralogues – Msg5 – is also redox-activated. We have also proposed a model for the mechanism of WH-phosphatases from budding yeast (Figure 77). Here the formation of a disulphide-bond between a cysteine in the amino-terminal domain of the protein with a conserved cysteine in the active site acts as a switch to activate these enzymes in oxidising conditions, when the activities of other MKPs may be compromised.
 |
|
Fig. 77: Proposed model for the activation of Sdp1 under oxidative conditions leading to Slt2 dephosphorylation. |
In recent years, redox-mediated reactions that influence and regulate signalling pathways within the cell have become increasing recognised as key events in many cellular processes. The discovery of this novel mechanism for attenuating MAPK signalling in yeast raises the exciting prospect that analogous mechanisms remain to be uncovered in higher eukaryotes.
Acknowledgements
This work was funded by GlaxoSmithKline and Cancer Research UK.
Principal publication and authors
G.C. Fox (a), M. Shafiq (b), D.C. Briggs (b), P.P. Knowles (b), M. Collister (b), M.J. Didmon (b), V. Makrantoni (b), R.J. Dickinson (b), S. Hanrahan (b), N. Totty (b), M.J.R. Stark (b), S. Keyse (b), N.Q. McDonald (b), Nature 447, 487–492 (2007).
(a) BM16, ESRF
(b) Cancer Research UK
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.