- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2006
- Surface and Interface Science
- Structure and reactivity of a model catalyst alloy under realistic conditions
Structure and reactivity of a model catalyst alloy under realistic conditions
Whereas most traditional electron or ion based surface science techniques are limited to vacuum environment, surface X-ray diffraction (SXRD) is able to determine structural parameters of surfaces under a pressure of several bars because of the negligible attenuation of hard X-rays by the reactant gas. The development of an in situ high pressure chamber for catalytic research at ID03 [1] has opened the door to an area in surface science, which has previously been impossible because of the so-called pressure gap between the conditions in an UHV experiment and those surrounding an operating industrial catalyst.
PtRh alloys can be used as “three-way” automotive exhaust gas catalysts. They have the ability to simultaneously remove unwanted, toxic exhaust gases such as CO, NOx and hydrocarbons. Gas interactions had been studied on PtRh surfaces under ultra high vacuum (UHV) conditions, but under realistic pressures, the behaviour of the alloy is in principle unexplored, in particular concerning the relationship between structure and reactivity.
Previous measurements under UHV conditions from the Pt25Rh75(100) had clearly indicated the formation of a well ordered ultra-thin RhO2 film at O2 pressures around 5 x 10-3 mbar using a sample temperature of 700 K. The resulting c(8 x 2) surface structure could be explained by an hexagonal RhO2 overlayer, or a so-called surface oxide [2]. The experimental observations were confirmed by DFT calculations and the resulting structure can be seen in Figure 92.
 |
|
Fig. 92: The atomic arrangement of the c(8 x 2) surface oxide tri-layer structure as found by UHV experiments and DFT calculations. Similar structures has been found on Rh(111), Rh(110) and Rh(100) [2]. |
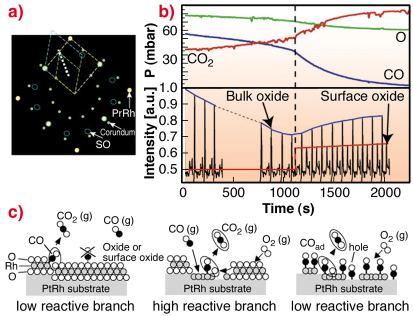
Because of electron based techniques being limited to UHV conditions, we turn to SXRD in order to study the surface phase under almost atmospheric gas pressures. The aim of these experiments was to approach conditions under which a real catalyst operates. A sketch of the in-plane reciprocal space in the presence of oxygen is shown in Figure 93a. Here the Pt25Rh75(100), the c(8 x 2) and the corundum Rh2O3(0001) bulk oxide reflections and their unit-cells are indicated.
By performing a scan in H and K as indicated in Figure 93a, we may detect the surface oxide as well as the bulk Rh2O3 corundum reflections at higher O2 pressures. Repetitive HK-scans as a function of time are shown in Figure 93b. In this way, we are able to connect our UHV observations with the SXRD studies at realistic conditions.
 |
|
Fig. 93: a) In-plane reciprocal space illustrating the unit cells of the Pt25Rh75(100) (yellow), the surface oxide (blue) and the Rh2O3 bulk oxide (green). b) Repetitive HK-scans according to the white dashed line in (a) for L = 0.5 during reaction conditions (bottom part) with simultaneous mass-spectroscopy measurements of the gas content (top part). c) Models of the surface phases and the CO-oxidation reaction process. |
To investigate the connection between surface structure and CO-oxidation reactivity, we simultaneously monitor the surface phases by SXRD and the gas composition by mass spectrometry, as shown in Figure 93b. As the CO is consumed by the oxidation into CO2, the partial CO/O pressure ratio is continuously reduced. When the CO pressure is sufficiently low, the non-bulk oxide covered surface reoxidises, which can be seen by the reappearance of the diffraction signal from the surface oxide. Concomitantly, the CO oxidation rate increases, as deduced from the measured partial pressures. Our observations lead us to conclude that under these conditions, the surface oxide film formed on the Pt25Rh75(100) surface is more active in the CO-oxidation than the corundum Rh2O3 bulk oxide and the metal surface on the Pt25Rh75(100) surface.
Although CO cannot adsorb on the surface oxide according to our DFT calculations (see Figure 93c), the oxygen stored in the film provides an additional source of oxygen expelled onto the metal surface, where it reacts with CO forming CO2. Under steady state conditions, the consumed surface oxide film is continuously regenerated by oxygen in the gas phase and consumed by the CO. The results provide confirmation in situ at the atomic scale and under realistic conditions of a general mechanism originally proposed by Turner in 1981 [3], and are likely to explain experimental observations on other late transition metal surfaces as well as on their corresponding nano-particles.
References
[1] M.D. Ackermann, et al., Phys. Rev. Lett. 95, 255505 (2005); P. Bernard, et al., Rev. Sci. Instrum. 70, 1478 (1999).
[2] E. Lundgren, et al., J. Phys. Cond. Matter 18, 481 (2006).
[3] J. E. Turner, B. C. Sales, M. B. Maple, Surf. Sci. 103, 54 (1981).
Principal Publication and Authors
E. Lundgren (a), J. Wang (b), M. Ackermann (c), R. Westerström (a), J. Gustafson (a), A. Resta (a), J. N. Andersen (a), A. Mikkelsen (a), O. Balmes (d), X. Torrelles (e), J. W. M. Frenken (c), B. Hammer (b), to be published.
(a) Department of Synchrotron Radiation Research, Lund University, (Sweden)
(b) Interdisciplinary Nanoscience Center (iNANO) and Department of Physics and Astronomy, University of Aarhus (Denmark)
(c) Kamerlingh Onnes Laboratory, Leiden University (The Netherlands)
(d) ESRF
(e) Institut de Ciencia de Materials de Barcelona (Spain).
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.