- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2005
- Surface and Interface Science
- "Live" Observation of Interplay between Structure and Chemical Activity of a Model Catalyst
"Live" Observation of Interplay between Structure and Chemical Activity of a Model Catalyst
Surface Science establishes a discipline within physics and chemistry that has evolved over the last 40 years to full maturity. Recently, the emphasis has been shifting away from static structures towards surface processes and dynamics. This is especially important in the light of the role that surface processes play in many practical phenomena and applications. A prominent example of such processes are chemical reactions at surfaces, i.e. heterogeneous catalysis.
The traditional surface-science approach to catalysis is to investigate clean, well-ordered, single-crystalline surfaces under highly idealised conditions of low temperatures and low pressures (UHV) and follow the interaction of pure gasses at low pressures with these surfaces. Much is known on regular adsorption structures and the energetics of adsorption, diffusion, reaction and desorption. The philosophy has been to extrapolate this data to high pressures and high temperatures. This requires an unrealistic leap of faith over the so-called ‘pressure gap’ of typically 10 orders of magnitude, separating these laboratory experiments and practical conditions. Compared to most UHV-based techniques, X-rays are almost completely unaffected by the presence of the gas phase. Thus, Surface X-Ray Diffraction (SXRD) can resolve the structure of the active catalyst under true operating conditions without any loss of accuracy. Such experiments bring the additional advantage of introducing realistic reaction rates that bring the concentrations of reaction products to a level that can easily be measured on-line, simultaneously with the diffraction measurements. This combination provides a one-to-one correlation between the atomic-scale structure and the catalytic activity of the catalyst surface.
A favorite model reaction system is the oxidation of CO on Pt. Recent high-pressure STM observations on Pt(110) have demonstrated that at a sufficiently high O2/CO pressure ratio this surface undergoes a structural phase transition, which has a dramatic effect on the reaction mechanism and strongly enhances the reaction rate. The STM data have been interpreted in terms of the formation of a thin platinum oxide film on the Pt(110) surface, on which the CO molecules oxidise through a Mars-Van Krevelen mechanism [1]. Other experiments as well as theory [2] strongly suggest a similar scenario on other metal surfaces.
 |
|
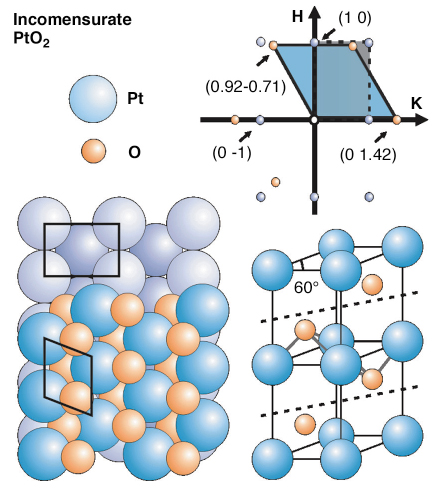
Fig. 105: Top right: in-plane reciprocal space map of the Pt(110) surface (blue circles) and the extra diffraction maxima due to the (PtO2) oxide layer; bottom: ball model of bulk terminated and oxide covered Pt(110) (left) and ball model of bulk |
Here we present the first observation by high-pressure SXRD performed at the ID03 beamline, of a correlation between the reactivity and the structure of a catalyst surface. The experiments performed during CO oxidation on Pt(110) show that the most active structure is a thin oxide film, namely PtO2. After preparing the surface under UHV conditions, the surface was exposed to 500 mbar of O2. This led to new diffraction peaks at non-integer H and K positions in reciprocal space, forming a regular, slightly distorted hexagonal pattern. The unit mesh of this Pt layer was very close to that of ![]() -PtO2, suggesting that the overlayer was a distorted PtO2 layer, azimuthally aligned with the substrate, and oriented with its c-axis along the surface normal (Figure 105). The oxide crystal truncation rods supported this interpretation and showed that the oxide was only 2 to 3 monolayers thick.
-PtO2, suggesting that the overlayer was a distorted PtO2 layer, azimuthally aligned with the substrate, and oriented with its c-axis along the surface normal (Figure 105). The oxide crystal truncation rods supported this interpretation and showed that the oxide was only 2 to 3 monolayers thick.
When exposed to CO, the oxide is reduced and we recover a metallic (CO covered) surface. In mixtures of CO and O2, the surface can switch between the oxide and the bulk-terminated metallic form.
 |
|
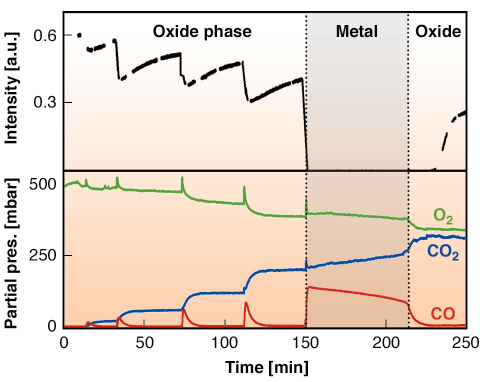
Fig. 106: Simultaneously measured X-ray diffraction intensity from the PtO2 oxide (top panel) and partial pressures of CO, O2, and CO2 (bottom panel). Separate CO pulses were admitted to the reactor, which was initially filled with 500 mbar of O2 at a temperature of 625 K. |
The full power of the method becomes apparent when we combine the diffraction experiment with simultaneous measurements of the CO oxidation reactivity (Figure 106). Starting with the ![]() -PtO2 surface in 500 mbar of O2, we admitted pulses of pure CO. The CO started reacting to CO2 as it entered the chamber. From the increase in the CO2 pressure, the typical turnover number can be estimated to be 3 x 103 molecules/site/s at 625 K in 80 mbar CO and 500 mbar O2.
-PtO2 surface in 500 mbar of O2, we admitted pulses of pure CO. The CO started reacting to CO2 as it entered the chamber. From the increase in the CO2 pressure, the typical turnover number can be estimated to be 3 x 103 molecules/site/s at 625 K in 80 mbar CO and 500 mbar O2.
The fifth CO pulse, at t = 150 min, was so large that the oxide layer was reduced completely: the diffracted intensity from the oxide layer dropped to zero and the surface reverted to the metallic state. Simultaneously, a strong decrease was observed in the reactivity, as well as a significant change in the reaction kinetics, i.e. the reaction mechanism.
The combined measurements of surface structure and reaction dynamics explicitly demonstrate that a surface oxide is formed on Pt(110) that, contrary to traditional ‘wisdom’, exhibits a significantly higher reaction rate than the original metallic surface.
Our observations show that in situ measurements under actual reaction conditions are an absolute necessity for a meaningful investigation of the complex behaviour of model catalysts.
The potential of high pressure in situ experiments is underlined by the attention and strong support they are presently receiving, for example within the ESRF (refurbishment of ID03) and within the European community (NanO2 project).
References
[1] B.L.M. Hendriksen et al, Phys. Rev. Lett. 89, 046101 (2002)
[2] K. Reuter, M. Scheffler, Phys. Rev. Lett. 90, 046103 (2003).
Principal Publication and Authors
M.D. Ackermann (a,b), T.M. Pedersen (c), B.L.M. Hendriksen (b), O. Robach (d), S.C. Bobaru (b), I. Popa (a), C. Quiros (e), H. Kim (a), B. Hammer (c), S. Ferrer (f), J.W.M. Frenken (b), Phys. Rev. Lett. 95, 255505 (2005).
(a) ESRF
(b) Kamerlingh Onnes Laboratory, Leiden University (The Netherlands)
(c) Interdisciplinary Nanoscience Center (iNANO) and Department of Physics and Astronomy, University of Aarhus (Denmark)
(d) CEA-Grenoble DRFMC / SI3M / PCM17 (France)
(e) Depto. de Física, Faculdad de Ciencias, Universidad de Oviedo (Spain)
(f) CELLS - ALBA, Universita Autònoma de Barcelona (Spain)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.