- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2004
- Surface and Interface Science
- Potential-dependent Surface Compression during Au Electrodeposition
Potential-dependent Surface Compression during Au Electrodeposition
Electrochemical metal deposition processes have been a subject of substantial basic research during the last few years, motivated by current technologies as well as future applications. To obtain a better fundamental understanding of electrodeposition processes the homoepitaxial growth of Au on Au(111) in aqueous electrolyte solutions was studied. This is particularly interesting due to the complex, potential-dependent in-plane structure of Au(111) surfaces. The Au surface structure was monitored in situ during growth by grazing incidence X-ray diffraction (GID), performed at beamline ID32 using a photon energy of 20 keV. In contrast to most previous diffraction studies of electrochemical interfaces, which were performed in a "thin layer geometry", a newly developed "hanging meniscus" transmission X-ray cell was used, where the beam passes through 5-6 mm of electrolyte solution. The minimised cell resistance and nearly unrestricted mass transport in this cell allows a combination of in situ surface X-ray diffraction studies of rapid structural changes with high quality electrochemical measurements.
First the potential-dependent Au(111) surface structure in Au-free solution was investigated. In accordance with previous results obtained in "thin layer geometry" [1] a hexagonal pattern around the integer crystal truncation rods is found at potentials negative of a critical value (for 0.1 M HCl at 0.1 V vs. a Ag/AgCl reference electrode), indicative for the well-known (px![]() 3) surface reconstruction (Figure 92a). p saturates at ~ 22 at the most negative potentials. Subsequently the electrolyte was exchanged with a solution containing 5-200 mM HAuCl4, resulting in the diffusion-limited electrodeposition of Au with deposition rates of 0.25 to 1 ML/min. As before, reconstruction peaks were found, however, shifted outwards by up to 20% (Figure 92b). This corresponds to a compression of the reconstructed surface layer down to p ~ 19 or a surface strain
3) surface reconstruction (Figure 92a). p saturates at ~ 22 at the most negative potentials. Subsequently the electrolyte was exchanged with a solution containing 5-200 mM HAuCl4, resulting in the diffusion-limited electrodeposition of Au with deposition rates of 0.25 to 1 ML/min. As before, reconstruction peaks were found, however, shifted outwards by up to 20% (Figure 92b). This corresponds to a compression of the reconstructed surface layer down to p ~ 19 or a surface strain ![]() = -p-1 = -5.3%, respectively. To date, a similar deposition-induced Au(111) surface compression has neither been reported in electrochemical environment nor under UHV conditions.
= -p-1 = -5.3%, respectively. To date, a similar deposition-induced Au(111) surface compression has neither been reported in electrochemical environment nor under UHV conditions.
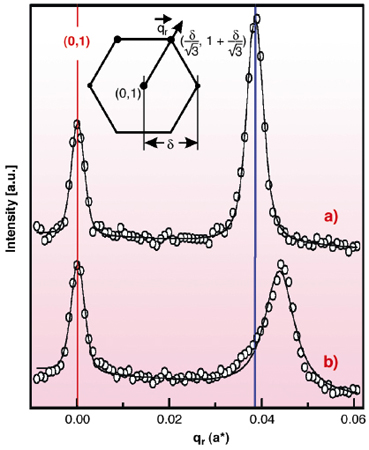 |
Fig. 92: X-ray scattering profiles along the |
Systematic studies revealed a pronounced potential-dependence of this effect (Figure 93). At potentials in the vicinity of the (px![]() 3)
3) ![]() (1x1) phase transition the surface compression is close to that in Au-free electrolyte (i.e., p ~22). p decreases approximately linearly with the potential E. This behaviour was observed in HCl, KCl, and H2SO4 solution, indicating that cation and anion species as well as HAuCl4 concentration (i.e. deposition rate) do not substantially influence the compression. To explain this phenomenon we suggest that the surface compression observed during Au deposition is closer to equilibrium in an electrochemical environment, whereas it is kinetically limited to p
(1x1) phase transition the surface compression is close to that in Au-free electrolyte (i.e., p ~22). p decreases approximately linearly with the potential E. This behaviour was observed in HCl, KCl, and H2SO4 solution, indicating that cation and anion species as well as HAuCl4 concentration (i.e. deposition rate) do not substantially influence the compression. To explain this phenomenon we suggest that the surface compression observed during Au deposition is closer to equilibrium in an electrochemical environment, whereas it is kinetically limited to p ![]() 22 in Au-free electrolyte. Using a simple thermodynamic model, the slope
22 in Au-free electrolyte. Using a simple thermodynamic model, the slope ![]()
![]() /
/![]() E can be rationalised by the potential-dependent change in surface stress, found at the Au(111) - electrolyte interface[2].
E can be rationalised by the potential-dependent change in surface stress, found at the Au(111) - electrolyte interface[2].
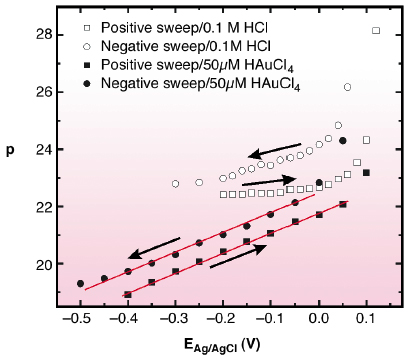 |
Fig. 93: Potential-dependent surface compression for the reconstructed Au(111) surface in 0.1M HCl (open Symbols) and 0.1M HCl + 50 µM HAuCl4 (solid Symbols). |
In summary, the potential dependent surface compression, observed in the presence of Au electrodeposition, may be viewed as a characteristic parameter of charged Au electrode surfaces, which may help to refine theoretical models of the reconstructed Au(111) surface and of metal surfaces in general, respectively.
References
[1] J. Wang, A.J. Davenport, H.S. Isaacs, B.M. Ocko, Science, 255, 1416-1418 (1991).
[2] C.E. Bach, M. Giesen, H. Ibach, T.L. Einstein, Phys. Rev. Lett., 78, 4225-4228 (1997).
Principal Publications and Authors
A.H. Ayyad, J. Stettner, O.M. Magnussen, Phys. Rev. Lett., in press.
Institut für Experimentelle und Angewandte Physik, Universität Kiel (Germany)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.