- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2004
- Structural Biology
- Investigating the Molecular Mechanism of Oncogenic Mutations of B-RAF
Investigating the Molecular Mechanism of Oncogenic Mutations of B-RAF
Cancer is a genetic disease in which changes in the DNA of a gene cause alterations in the properties (or amounts) of the encoded protein, disrupting its normal function in the regulatory processes that control the homeostasis and development of a healthy cell. The Cancer Genome Project is a systematic effort to identify all the genetic abnormalities that contribute to the development of cancer.
In the first stage of this genome-wide screen for mutated genes, Futreal and colleagues identified somatic mutations in the B-RAF gene [1]. This discovery was significant because B-RAF encodes a serine/threonine protein kinase that is a critical component of a signalling pathway (the MAP kinase pathway) that promotes cell growth and proliferation. In healthy cells, this is activated only in response to growth signals. Furthermore, the gene encoding the RAS protein, which activates normal B-RAF, is itself mutated to an oncogene in 15% of human cancers. B-RAF mutations are particularly prevalent in cutaneous malignant melanoma, being associated with 60% of such tumours, and also occur with moderate to high frequency in colorectal, ovarian and papillary thyroid carcinomas. These findings implicate activating oncogenic mutations of B-RAF as critical promoters of malignancy. Since the initial discovery [1], extensive analysis of the B-RAF gene associated with human cancers has identified over 30 single site missense mutations, mostly located within the kinase domain, clustered to two regions; the glycine-rich P-loop of the N-lobe, and the activation segment. Significantly, a glutamate (E) for valine (V) substitution at residue 599 in the activation segment accounts for 90% of B-RAF mutations in human cancers. The V599E mutant of B-RAF possesses the hallmarks of a conventional oncogene. The kinase activity of this mutant protein is elevated greatly, it constitutively stimulates the MAP kinase pathway in vivo independent of RAS, and potently transforms NIH3T3 cells. Similarly to the V599EB-RAF mutant most other, although significantly not all, B-RAF mutants that have been characterised biochemically, have elevated kinase activity.
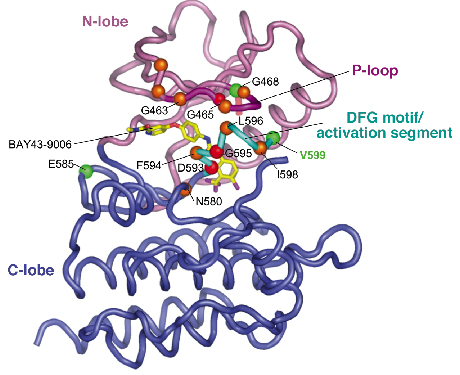 |
Fig. 74: Overall view of B-RAF kinase domain showing position of mutations (green, orange and red spheres, for high, intermediate and lower activity mutants) located at the P-loop/activation segment interface. |
To understand how activating mutations alter the conformation of B-RAF, and hence stimulate its kinase activity, we crystallised the B-RAF kinase domain and determined its structure with X-ray diffraction data collected at the microfocus beamline (ID13). The protein was crystallised in complex with a small molecule inhibitor of B-RAF termed BAY43-9006 developed by Bayer pharmaceuticals, and which is currently undergoing phase III clinical trials. Our structural information revealed a unifying hypothesis for the activation of B-RAF kinase activity by diverse B-RAF oncogenic mutations, and provided the first quantitative insights into the mechanism of inhibition of B-RAF by BAY43-9006. The structure we solved of normal B-RAF showed that the kinase domain adopts an architecture characteristic of other members of the protein kinase family, containing two lobes termed the N- and C-lobes (Figure 74). Kinases are regulated by changes in their conformation, and the structure of B-RAF revealed that the kinase domain adopted an inactive conformation, being incapable of binding ATP and catalysing MEK phosphorylation. The protein was inactive because a key regulatory region of the enzyme, termed the activation segment, was held in an inactive conformation as a result of hydrophobic interactions with the P-loop. Significantly, the regions of the activation segment and P-loop that interact with one-another, restraining the activation segment in an inactive conformation, are precisely where the majority of B-RAF oncogenic mutations are clustered. For example, the hydrophobic side chain of valine 599 of the activation segment contacts the side chain of phenylalanine 467 of the P-loop. This indicated that as a result of B-RAF mutations the inactive B-RAF conformation is destabilised thereby promoting an active B-RAF conformation. In addition, we proposed that a glutamate residue at position 599 (accounting for 90% of B-RAF mutations) promotes the active conformation by forming favourable electrostatic interactions with a lysine side chain of the aC-helix of the N-lobe (Figure 75).
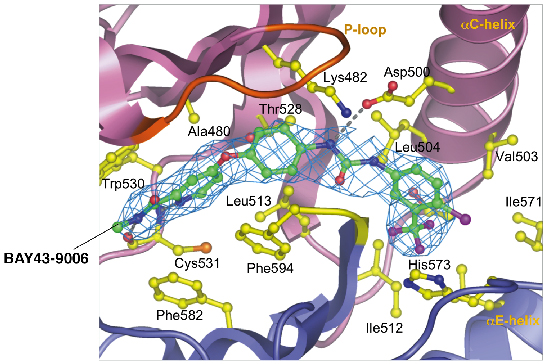 |
Fig. 75: Close-up view of BAY43-9006 bound to B-RAF. |
Our structural studies of B-RAF provide new intellectual insights for drug development. In future work we plan to use the structure of B-RAF to develop more specific inhibitors of the kinase, and also to explore the conformations of mutant activating forms of the kinase.
References
[1] H. Davies et al., Nature, 417: 949-954 (2002).
[2] P.T.C. Wan, M.J. Garnett, S.M. Roe, S. Lee, D. Niculescu-Duvaz, V.M. Good, C.M. Jones, C.J. Marshall, C.J. Springer, D. Barford, and R. Marais, Cell, 116: 855-867 (2004).
Authors
P.T. Wan, S.M. Roe and D. Barford,
Section of Structural Biology, Institute of Cancer Research, Chester Beatty Laboratories, London (UK)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.