- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2004
- Materials Science
- X-ray Powder Diffraction Study of Hexagonal Turkey Egg-white Lysozyme
X-ray Powder Diffraction Study of Hexagonal Turkey Egg-white Lysozyme
Obtaining the atomic structure of a large macromolecule like a protein depends upon the availability of good quality single crystals, the growth of which entails considerable skill and dedication. The conditions for optimal crystal growth must be painstakingly investigated, and a microcrystalline powder often precipitates instead of the sought-after crystal. Following the recent reports of crystal structure refinement [1] and solution [2] of some small proteins from powder diffraction data, the notion of obtaining at least some structural data from the powder looks potentially attractive, especially given the very high quality powder-diffraction patterns that can be measured at a modern synchrotron-radiation source. Despite all efforts, some materials of interest appear stubbornly to resist forming single crystals, the availability of the powder technique thus widens the spectrum of samples which might be investigated. Moreover, powder diffraction measurements can also give a range of complementary information beyond that which can be obtained from a single crystal. For example, the peak shapes depend on the microstructure of the material, accurate unit cell parameters can easily be determined, and the sample generally survives under more varied or extreme conditions. Here we report on our attempts to apply powder diffraction techniques to simple proteins.
In the present study, the structure of turkey egg-white lysozyme (TEWL) has been refined from high-resolution X-ray powder diffraction data. The sample was obtained in a polycrystalline form via rapid precipitation at high protein concentration using a 0.5 M NaCl solution (pH = 6) and the diffraction data were collected at room temperature employing the High-resolution Powder Diffraction (ID31) and the Materials Science (ID11) beamlines. Our structural analysis was initiated with a structural model that had previously been derived from single crystal data (PDB entry: 1TEW). Molecular replacement was shown to give a suitable starting point for refinement, illustrating that powder data can be sufficient for this approach. Crystallographic models were then refined by combined Rietveld and stereochemical restraint analysis of the powder data (dmin = 3.35 Å) resulting in the extraction of reliable lattice parameters and the refinement of the molecular conformation (Figure 32). The structure is hexagonal (Sp. group: P6122; a = 71.0945(3) Å, c = 85.0371(5) Å) with 12 symmetry related molecules in the unit cell, in agreement with previous studies.
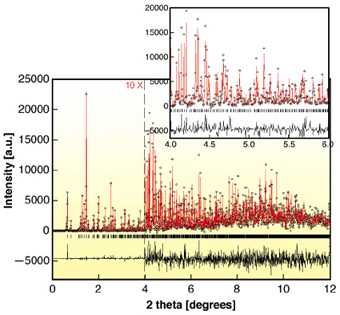 |
|
Fig. 32: The Rietveld fit for sample crystallised at pH 6 and 295 K ( |
TEWL is a relatively compact structure, comprising 8 helices and 4 anti-parallel ß-sheets. The overall and secondary structural topology of the powder structure are essentially the same as in the previously determined single-crystal structures with a few slight differences mainly in the regions of the flexible loops and the C and N-termini of the molecule. The average RMS deviation of the Ca positions, after aligning the original (PDB entry: 1TEW) and refined structures, was determined to be 0.95 Å over 128 residues. The deviation between the structures may arise in part due to the differences in the structural resolution of the data (dmin = 1.65 Å for 1TEW and 3.35 Å for the powder structure). The Ramachandran plot for the main-chain torsion angles (![]() ,
,![]() ) indicated that there were no torsion angles falling in disallowed regions.
) indicated that there were no torsion angles falling in disallowed regions.
Low temperature experiments indicated how strategies for cryocooling and cryoprotecting samples might be more easily developed with powdered samples. In this case, we have observed a significant contraction in TEWL upon freezing (Figure 33). Interpretation of the very low-resolution data obtained below the freezing point of the mother liquor would seem to indicate that the channels in the structure expand significantly and the protein becomes packed much more closely together.
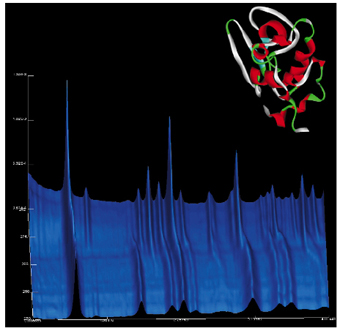 |
|
Fig. 33: Diffraction profiles plotted as a function of temperature (vertical axis) collected while warming the TEWL sample through the freezing transition at beamline ID11. Inset: Refined conformation of TEWL (pH 6.0) at RT illustrated as ribbon. |
These results illustrate that powder diffraction methods can provide a wealth of complementary information about the structure of protein crystals. Molecular replacement techniques can be used to find out how the molecule packs into a unit cell for a particular structural modification. The molecular structure may also be refined by using stereochemical restraints in addition to the usual Rietveld method for powder profile refinement.
References
[1] R.B. Von Dreele, J. Appl. Cryst. 32, 1084-1089 (1999).
[2] R.B. Von Dreele, Acta Cryst. D56, 1549-1553 (2000).
Principal Publication and Authors
I. Margiolaki, (a), J.P. Wright (a), A.N. Fitch (a), G.C. Fox (a) and R.B. Von Dreele (b), Acta Cryst. D, accepted.
(a) ESRF
(b) ARGONNE, IL 60439 (USA)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.