- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2003
- Materials Science
- Global Structure Kinetics of Photochemical Reactions in Solution
Global Structure Kinetics of Photochemical Reactions in Solution
It is well known that chemical reactions in solution are strongly determined by the role of the solvent as reaction barrier and energy reservoir. The photoreaction of iodine in solution has long been the model system of choice for the study of reaction kinetics. In particular, I2 in CCl4 is very important due to its reduced-rate kinetics [1].
A method has been developed to resolve the structural dynamics of a photoexcited system by pulsed X-ray scattering, that allows a natural time resolution of 100 ps. Stroboscopic data acquisition is achieved by synchronising a femtosecond laser to the chopped X-ray pulses from an in-vacuum undulator at ID09 using its full emission spectrum at 3% bandwidth. The high flux of 1011 photons per second at 900 Hz pulse rate is indispensable regarding the low photo induced modulation signals of the full scattering in the intensity range of 10-4 [2].
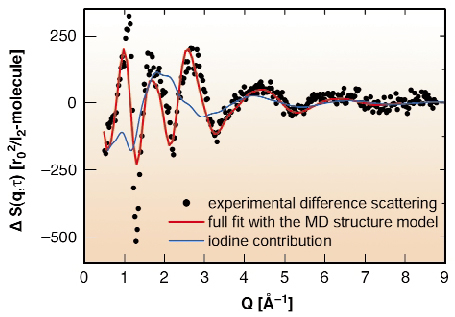
The information content differs depending on the region in q space considered (see Figure 63). At high momentum transfer, where strongly correlated atomic structures manifest, the distance changes of the iodine atoms can be observed. A mixture of states is observed within the first 2 nanoseconds, the stretched molecule (so-called A/A' state) and iodine atoms from the dissociation reaction. Occupancies and lifetimes can be inferred from the time-resolved q scans.
 |
|
Fig. 63: Difference in scattering cross section in between the nonexcited sample and 1 ns after the excitation of the iodine absorption band by femtosecond laser pulses at 519 nm wavelength. The red line represents the model data derived by molecular dynamics simulations of the complete system of iodine within the CCl4 solvent using time resolved scattering theory. The blue line shows only the local signal change of iodine and its solvation cage. |
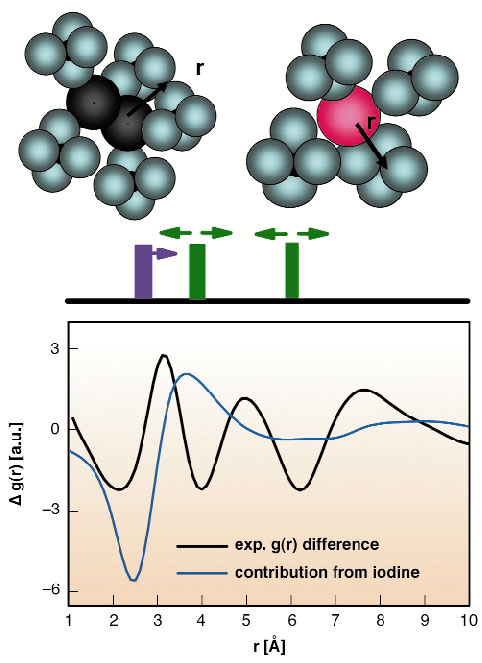
New, subtler changes manifest in the low q-region, where long range distances and liquid structures are observed. There, the solvent makes a large contribution to the signal due to the heat released by the chemical reaction. Part of the signal also arises from the changes in the pair distribution function of iodine to adjacent Cl atoms within the solvation cage and the outer shells. The collapse of the cage around a single iodine atom causes the solvent to come closer to an iodine scattering centre, whereas the extended I2 state pushes the solvent outwards. The sum of all contributions is seen in Figure 64, which represents the Fourier inversion of the difference scattering data. The interpretation is confirmed by molecular dynamics (MD) studies (red line in Figure 63). For this purpose a MD simulation has been performed with a box of 256 CCl4 molecules and one iodine molecule using appropriate pair potentials and steps of 1 fs in a 1ns observation interval.
 |
|
Fig. 64: Change in the pair distribution function obtained by Fourier inversion of the difference scattering in Figure 63 (black line). Again the blue line marks the contribution from iodine and its cage. The signal can be understood as a change of intermolecular distances of the iodine (magenta) in the excited A/A' state and distance changes between chlorine atoms of CCl4 (green), which shift both to lower and higher distances, giving rise to the observed oscillations (black curve). |
At larger time delays hydrodynamics comes into play as the energy flow from the excited iodine centres to the solvent is completed and the pressure in the excited region relaxes. At about one microsecond the system is equilibrated, both thermally and mechanically, with recombined iodine molecules and the heated solvent, whose difference scattering signal persists. Thus the solvent acts as a calorimeter that mirrors the energy deposited into the system by the photoreaction. This energy can be compared to the number of excited iodine molecules to give information about ultrafast geminate reactions beyond the time resolution.
The challenge for the future will be to extend the procedure to larger molecules and biologically relevant aqueous solutions of photoactive molecules.
References
[1] A.L. Harris, J.K. Brown and C.B. Harris, Ann. Rev. Phys. Chem. 39, 341-366 (1988).
[2] M. Wulff, A. Plech, L. Eybert, R. Randler, F. Schotte and P. Anfinrud, Faraday Disc. 122, 13-26 (2002).
Principal Publication and Authors
A. Plech (a), M. Wulff (d), S. Bratos (b), F. Mirloup (b), R. Vuilleumier (b), F. Schotte (c) and P. Anfinrud (c), accepted in PRL.
(a) Fachbereich Physik der Universität Konstanz Germany)
(b) Université Pierre et Marie Curie, Paris (France)
(c) National Institutes of Health, Bethesda (USA)
(d) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.