- Home
- Users & Science
- Scientific Documentation
- ESRF Highlights
- ESRF Highlights 2000
- Chemistry
- Following Transient Structural Changes in Organic Solids
Following Transient Structural Changes in Organic Solids
With common optical pump-probe techniques it is possible to follow structural relaxation processes provided they coincide with spectral changes. However, experimental results obtained with these techniques can only be interpreted when the potential energy surface (PES) of the (transient) species is well known and understood. For this, time-dependent classical and quantum-chemical calculations are carried out and the results are compared with experimental data. Since time-resolved X-ray diffraction directly probes the structural changes (via reciprocal space), no information about the PES is needed. This method gives detailed information about the structural rearrangements without prior knowledge of the strongly perturbed PES, especially in systems where the photo-physical relaxation pathway is dominated by large-amplitude motions.
In the following, systems will be discussed where the photo-physical behaviour is strongly coupled and influenced by large-amplitude motions. The time dependence of the structural changes is studied by optical pump X-ray probe schemes at ID9-TR. With this technique it is possible to follow the structure factors of a system as a function of time. After photo-initiation of the reaction by a 300 fs optical laser pulse, the excited state is probed with a 150 ps X-ray pulse [1]. By varying the delay between pump and probe, a series of snapshots of the moving structure can be taken, where each shot probes the average structure (non-excited and excited, including the dispersion of the latter) at a given time. Since the typical lifetime of the systems reported here is in the nanosecond range, this is equivalent to following the structural relaxation pathways during the lifetime of the excited state on the excited-state PES.
We have studied the time-resolved (TR) powder diffraction of N,N-dimethylaminobenzonitrile (DMABN, C9H10N2, Figure 10), a compound widely discussed in the literature with emphasis on its relaxation mechanisms during photo-excitation [2], and a special case of a molecular charge-transfer compound. Our aim was to measure the structural relaxation following photo-excitation and to test the feasibility of using this method to determine displacements at atomic resolution on a picosecond timescale. One of the main questions was whether structural relaxation takes place in a crystal and, if so, what is the amplitude and timescale (Figure 10). With TR X-ray diffraction, it should be possible to distinguish between the inversion motion of the CH3-groups around the N-atom and the torsional motions of the CH3-groups as relaxation processes on the excited state PES. Depending on the kind of motions, electronic-coupling effects between the ![]() electrons (torsion) or vibronic coupling as in NH3 (inversion) characterise the photo-physical behaviour of DMABN.
electrons (torsion) or vibronic coupling as in NH3 (inversion) characterise the photo-physical behaviour of DMABN.
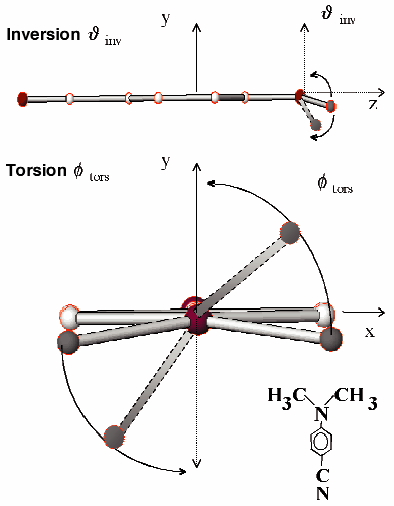 |
Fig. 10: The intramolecular degrees of freedom, which contribute to the relaxation process (inversion
|
The results obtained from monochromatic TR X-ray diffraction are presented in Figure 11. Figure 11a summarises the complex relaxation behaviour of one asymmetric DMABN unit with respect to the coupled motions, torsion and inversion, as a function of time. In Figure 11b the time decay of the averaged torsional angle is plotted. Not shown in the picture is the change of the inversion from 13° to 3° and back, which essentially follows the same time dependency.
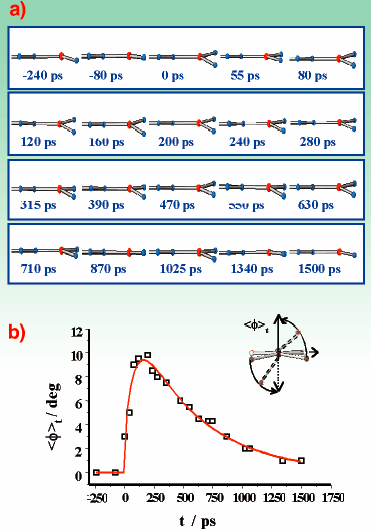 |
Fig. 11: Relaxation pathway of light-excited DMABN as a function of time: a) Change of inversion and torsion of an asymmetric unit as a function of time; b) Time-dependence of the averaged torsional angle <
|
References
[1] F. Schotte, S. Techert, P. Anfinrud, V. Srajer, K. Moffat and M. Wulff in Handbook of Synchrotron Radiation, Vol. 5, Ed. D. Mills, Wiley and Sons (2000).
[2] C. Chudoba, A. Kummrow, J. Dreyer, J. Stenger, E.T.J. Nibbering, T. Elsaesser and K.A. Zachariasse, Chem. Phys. Lett., 309, 357 (1999).
Principal Publication and Authors
S. Techert (a, b), F. Schotte (a) and M. Wulff (a), Phys. Rev. Lett., 86(9), (2001).
(a) ESRF
(b) Max-Planck-Institute for Biophysical Chemistry, Göttingen (Germany)
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.