Materials
Introduction
The detailed structure of crystalline materials at the atomic level is obtained in a well-established way by X-ray diffraction. Under ordinary circumstances a standard diffractometer with an ordinary X-ray tube can solve the problem. There are, however, cases in which sufficiently large crystals cannot be grown, powder diffraction patterns cannot be resolved due to too low angular resolution or structures are not periodic as occurs with quasicrystals. In many of these cases, synchrotron radiation can provide the means to overcome the problems.
Microcrystal diffraction from a novel magnesium aluminophosphate
Microporous materials with cage structures such as zeolites have found extensive industrial use over the last decades. They serve as catalysts, ion exchangers and gas absorbers, and have many other applications. Their function is often correlated with their large internal surface area. The silicon-aluminium based zeolites can be found in nature but recently zeolite analogues with other tetrahedral atoms such as phosphorous, nickel, cobalt etc have been synthesised. The molecule-selective properties of these compounds depend on their cage structures where the ring openings are determined by the 8, 10, 12 or higher order tetrahedral atom-oxygen atom rings. These compounds are formed by hydrothermal synthesis of gels of various compositions. In this case aluminium hydroxide, magnesium acetate, phosphoric acid and an organic template molecule were used. In the hydrothermal synthesis, these gels are heated under pressure for days or weeks and the final product is, in general, a microcrystalline product. Each produced material has a unique combination of channels and cages, and therefore possesses individual material properties. Recently, organic bases and alkylammonium ions have been used as structure-directing agents, or templates, for crystallisation of microporous materials. In this study the use of templates has been further extended by using bulky diquinuclidic templates of the general formula (C7H13N)-(CH2)n-(C7H13N) [1]. Figure 51 illustrates the molecule with n = 4 used in this case. These materials, however, rarely produce crystals larger than a few tens of microns and the precise structural characterisation by single crystal diffraction is usually impossible using conventional X-ray sources. In the past, powder diffraction has been the method of choice but with the increasing complexity of the structures, the solution of the structures has proven difficult. In this study, early powder diffraction studies failed to produce a solution. Here we demonstrate the potential of microcrystal diffraction at the Materials Science beamline, ID11, with its extremely high flux and crystallographic infrastructure. A crystal of approximate size 0.03 mm on edge was used for a data collection at a wavelength of 0.512 Å using a Siemens SMART CCD data collection system. The crystal was cooled to 200 K and 7925 reflections were collected in a few hours. The structure solution in space group R-3 with a cell a = b = 12.760 (2), c = 30.939 (6) Å revealed one of the most complex aluminophosphate structures to date. The large cages housing the bulky template are elongated along the crystallographic c axis (see Figure 52).
The precise structure analysis allowed the location of the template in the large cages, which is one of the first times that the relation between template and framework could be established in detail. This information will give clues to further crystal engineering attempts in designing other catalysts. Figure 53 illustrates the fit of the template in the cage. This compound can be cleared of its template by heating to + 500 °C. The calcined compound can then be used as a catalyst for reactions of methanol to light olefins.
Publication
G.W. Noble (a), P.A. Wright (a), P. Lightfoot (a), R.E. Morris (a), K.J. Hudson (a), Å. Kvick (b), H. Graafsma (b), Angew. Chem. Int. Ed. Engl. 36, 81 (1997).
(a) School of Chemistry, University of St. Andrews (UK)
(b) ESRF
Crystal structure solution from powder diffraction data
To understand the properties of solid materials, knowledge of the crystal structure is essential. The crystal structure gives a detailed picture of the arrangement of the atoms and molecules making up the solid, which is highly regular for crystalline materials. The routine method to determine a crystal structure is to use single-crystal diffraction techniques. Many compounds, however, are not conveniently obtained as single crystals, but are in the form of microcrystalline powders. Solving a crystal structure from powder data is more difficult than from a single crystal, because all the three-dimensional information present with a crystal is hidden by the random orientations of the crystallites making up the powder. The overlap of adjacent diffraction peaks may become so severe for structures with large unit cells or of low symmetry, that the individual peak intensities and peak positions are lost. To solve a structure from powder data, therefore, requires high quality diffraction data, with narrow peak widths to reduce peak overlap, and free from systematic errors. Synchrotron radiation is well suited for this, and the powder diffractometers on the ESRF beamline BM16 and the Swiss-Norwegian beamline (SNBL) BM1 produce ideal data. A number of problems have been solved that represent an advance in the complexity of structural problems that can be solved from powder-diffraction data.
For example, the compound (CH3)2SBr2 is known to exist in a stable and a metastable forms. The two forms were believed to correspond to a covalent charge-transfer compound ((CH3)2S![]() Br2) and an ionic compound ((CH3)2SBr+Br-), respectively, as deduced from vibrational (infrared and Raman) spectroscopy. The structures were unknown. A powder diffraction pattern of each compound was collected on the SNBL. The stable form was rapidly solved, owing to the high quality of the data [1], but it was very difficult to determine the unit cell of the metastable material using standard indexing programs. The pattern was eventually indexed, but as a mixture of two distinct phases, with different compositions. One phase is orthorhombic, with a unit cell volume of 2720 Å3, and the other is monoclinic with a cell volume of 937 Å3. The crystal structures were solved using Direct Methods (program SIRPOW). The orthorhombic phase corresponds to an ionic form, but with additional bromine molecules in the structure whose presence was not anticipated, Figure 54. The monoclinic phase is a charge-transfer form, but distinct from the stable form, as the structure also contains additional bromine atoms. Only a crystal structure determination is able to reveal the true nature of this sample. Hence two quite complex structures have been solved from one diffraction pattern [2]. Both phases are of excellent crystallinity, and there is nothing in the width or shape of the observed peaks that suggest which peaks arise from which phase, or indeed how many phases are present. Only by having data of the highest quality could such a problem be solved.
Br2) and an ionic compound ((CH3)2SBr+Br-), respectively, as deduced from vibrational (infrared and Raman) spectroscopy. The structures were unknown. A powder diffraction pattern of each compound was collected on the SNBL. The stable form was rapidly solved, owing to the high quality of the data [1], but it was very difficult to determine the unit cell of the metastable material using standard indexing programs. The pattern was eventually indexed, but as a mixture of two distinct phases, with different compositions. One phase is orthorhombic, with a unit cell volume of 2720 Å3, and the other is monoclinic with a cell volume of 937 Å3. The crystal structures were solved using Direct Methods (program SIRPOW). The orthorhombic phase corresponds to an ionic form, but with additional bromine molecules in the structure whose presence was not anticipated, Figure 54. The monoclinic phase is a charge-transfer form, but distinct from the stable form, as the structure also contains additional bromine atoms. Only a crystal structure determination is able to reveal the true nature of this sample. Hence two quite complex structures have been solved from one diffraction pattern [2]. Both phases are of excellent crystallinity, and there is nothing in the width or shape of the observed peaks that suggest which peaks arise from which phase, or indeed how many phases are present. Only by having data of the highest quality could such a problem be solved.
In another example, synchrotron powder diffraction patterns were collected at Daresbury Laboratory, and on SNBL and BM16 at the ESRF in a joint effort to solve the unknown structure of fluorescein diacetate [3]. This is a relatively complex organic compound with 31 carbon and oxygen atoms (C24H16O7), making up a total of five linked ring systems in the molecule. The structure is triclinic and was solved from data collected at low temperature on BM16, using Direct Methods (SIRPOW again). As well as the triclinic crystal structure, the program solved the complete molecular structure, with all 5 ring systems, and every single C and O atom correctly assigned, Figure 55. The bond distances in the molecule correspond to within 0.1 Å of those found in similar molecules, even though this was not included as prior information. This represents to our knowledge the most complex structure of an organic molecule solved by Direct Methods from powder-diffraction data, and suggests that even larger systems may be accessible. This is of obvious interest for characterising organic molecules with pharmaceutical applications, where knowledge of which polymorph is formed is of vital importance as this can affect the physical properties such as rate of dissolution, solubility, etc.
Publication
[1] A.J. Mora (a, g), A.N. Fitch (b), P.N. Gates (c) and A. Finch (c), in "European Powder Diffraction Conference: EPDIC IV", Materials Science Forum, 228, 601, (1996).
[2] G. Vaughan (b), A.J. Mora (a, g), A.N. Fitch (b), P.N. Gates (c) and A. Finch (c) , to be published.
[3] K. Knudsen (d), P. Pattison (d, e), A.N. Fitch (b) and R.J. Cernik (f), to be published.
(a) Department of Chemistry, Keele University (UK)
(b) ESRF
(c) Department of Chemistry, Royal Holloway and Bedford new College, Egham (UK)
(d) Swiss Norwegian beamline, ESRF
(e) Dept of Crystallography, University of Lausanne, (Switzerland)
(f) Daresbury Laboratory, Warrington (UK)
(g) Departamento de Quimica, Universidad de Los Andes, Merida (Venezuela)
Phason disorder in icosahedral AlPdMn quasicrystals
Quasicrystals are highly-ordered structures with no translational symmetry. They can be obtained as large single grains and present an icosahedral symmetry. Because of the lack of translation invariance, a new kind of defects named phason can be present in the structure. At the atomic scale it corresponds to the possibility for an atom to occupy two neighbouring positions with the same local environment. The sharp Bragg reflections are indexed with 6 integer indices. The 6-D reciprocal lattice vector Q decomposes in two 3-D vectors Qpar and Qper where Qpar is the reciprocal vector in physical space and Qper the component in the complementary space. The possible correlation between the phason defects leads to different effects in the diffraction spectrum, but the relevant vector in reciprocal space to be considered is Qper. Although centimetre size single grain are obtained in the AlPdMn system, two kinds of phason defects have been observed on D2AM (BM2).
A distribution of phason strain was observed by measuring Bragg reflections profile in a broad Qper range for two samples, one mechanically polished and the other one obtained by cleavage. Although low Qper reflections are extremely sharp, there is a clear dependence of the width of the reflection with Qper for both samples demonstrating the presence of a linear phason strain field in the sample. It has to be noticed that the high Qper reflections are extremely weak, and that a dynamical range of more than 8 orders of magnitude was necessary to carry out this experiment. When comparing the polished and the cleaved sample, a dramatic difference shows up: the Bragg peak width of the polished sample increases 2.5 times faster as a function of Qper than for the cleaved sample (Figure 56). This demonstrates that phason strain has been introduced by mechanical polishing.
A weak diffuse scattering was also observed around the Bragg reflections. The intensity distribution is anisotropic, as shown in Figure 57 (top) where isointensity in the (Qx, Qy) plane is presented around two symmetry-equivalent reflections. This distribution is well reproduced using the elasticity theory of quasicrystal [Jaric et al.] and only 2 phasons elastic constants as parameter (Figure 57 bottom), as previously observed with elastic neutron scattering [de Boissieu et al.]. This weak diffuse scattering is thus the result of frozen in long wavelength phason fluctuations, specific to quasicrystal.
Besides the diffuse scattering around the Bragg reflections we also observed streaks of diffuse scattering elongated along the 3-fold directions. They have a cylindrical shape, with a FWHM of around 0.1 Å-1, and seem to be related to the existence of a phase transition for a chemical composition slightly different from the one of the icosahedral phase. The interpretation of the origin of this diffuse scattering is underway.
Publications
[1] M. Boudard (a), M. de Boissieu (a), J.P. Simon (a), J.F. Berar (b), B. Doisneau (a), Phil. Mag. Lett, (1996), 74 429
[2] M. de Boissieu (a) , M. Boudard (a) , A. Letoublon (a), J.P. Simon (a) , J.F. Berar (b), to be published.
(a) LTPCM, CNRS, INPG, UJF, St Martin d'Hères Cedex (France)
(b) Laboratoire de Cristallographie, CNRS, Grenoble (France)
Diffraction Anomalous Fine Structure (DAFS) study of epitaxial semiconductors
Strained pseudobinary III-V semiconductors are of great interest due to their applications in the field of electronic and optoelectronic devices: the presence of strain reduces the symmetry of the crystal and modifies the electronic band structure. It is also known that the performances of the epitaxially grown heterostructures are affected by the composition gradient at the interfaces. Therefore efforts are being made to determine the local strain through the whole heterostructures as well as the atomic structure at the interfaces. The XAFS (X-ray Absorption Fine Structure) spectroscopy cannot be applied to study the local environment in a straightforward way due to the peculiar nature of the epitaxial samples: too thin to be measured in transmission and often grown on a substrate having some of the atomic components in common with the epilayer. Glancing-angle EXAFS or SEXAFS have been used but they solve the problem only in part, since the signal collection is restricted to a few monolayers below the surface, allowing only quasi-surface measurements. The DAFS spectroscopy is an alternative probe: it combines the local structural sensitivity of XAFS with the long range crystallographic sensitivity of X-ray diffraction by measuring Bragg peak intensities as a continuous function of energy through an absorption edge. The energy dependent modulation of the peak intensity contains local structural information similar to that of XAFS. The advantage of DAFS for studying this kind of systems, is to give structural information about the whole heterostructure by choosing the Bragg peaks (site-selective Bragg peak) of the strained phase. The site selectivity of DAFS is also used for studying the interfaces.
The raw DAFS spectra at the Ga and As K-edges (Figure 58), measured at the CRG-D2AM beamline (BM2) with the (006) reflection of a 5000 Å thick GaAs1-xPx epilayer (x = 0.225), show an energy resolution of less than one eV and a signal-to-noise ratio better than 0.5%. The crystallographic-based co-refinement of the two spectra gave an As concentration of 0.78 and showed that the calculated intensities are very sensitive to small atomic displacements (~ 0.01 Å). The XAFS-like signals were extracted and normalised with parameters obtained from the crystallographic structure: Figure 59 shows the Fourier transforms. A standard EXAFS data analysis was then performed.
The data analysis is still in progress, however, two results can be drawn from the study of GaAs1-xPx epilayers with different amounts of strain: a) the first nearest neighbour Ga-As distance slightly increases (2.40 to 2.43 Å) with increasing residual built-in strain (0.4% to 0.6%), this is confirmed by the second shell Ga-Ga distance (3.94 to 3.98 Å), b) the Ga-P distance is almost not affected (2.36 to 2.37 Å) by the strain, confirming the results of the Raman measurements performed on the same samples. The results demonstrate the possibilitiy of obtaining high quality DAFS spectra at the ESRF and show that the DAFS spectroscopy can give precise XAFS-like and crystallographic information.
Publication
[1] M. G. Proietti (a), H. Renevier (b), J. F. Bérar (a), V. Dalakas, J. L. Hodeau (a), G. Armelles (c), J. Garcia (a), J. Physique IV, (1997).
[2] M.G. Proietti (a), H. Renevier (b), J.F. Bérar (b), J.L Hodeau (b), G. Armelles (c), and J. García (a), to be published.
(a) Instituto de Ciencia de Materiales de Aragón, CSIC- Universidad de Zaragoza (Spain)
(b) Laboratoire de Cristallographie, CNRS-UJF, Grenoble (France)
(c) Instituto de Microelectrónica de Madrid, CSIC, Madrid (Spain)
Temperature dependence of non-Debye disorder in doped manganites
Hole-doped manganites have attracted a great deal of scientific interest, initially triggered by the observation of a colossal negative magnetoresistance (CMR). However these materials have also a more fundamental interest for their delicate balance and interplay between electronic, magnetic and structural properties. It is generally accepted that Zener's double exchange mechanism is at the root of the unusual properties of these materials; however the simple double exchange hamiltonian cannot account for many qualitative and quantitative aspects of the properties of these materials, such as the observed behaviour of the metal-insulator (MI) transition as a function of temperature. To explain such behaviour, an electron-phonon interaction term arising from an assumed dynamical Jahn-Teller distortion of the MnO6 octahedra was added to the double exchange hamiltonian.
The presence and role of such additional disorder in modifying the unusual properties of these CMR materials in general and metal-insulator transitions in particular has been investigated on the CRG beamline GILDA (BM8) at the ESRF using the EXAFS (Extended X-ray Absorption Fine Structure) technique. For this purpose the La1-xCaxMnO3 system was chosen, as the metal-insulator transition in this series has been well characterised: the metal-insulator transition temperature can be systematically varied by changing x and the transition temperature can be substantially modified even for a fixed x by varying the heat treatment. Samples with x = 0.2 and 0.4 were investigated as a function of temperature and preparation method. The EXAFS results clearly showed an increase of non-Debye disorder at the MI transition temperature significantly correlated with the resistivity behaviour (Figure 60).
Publication
C. Meneghini (a, b), R. Cimino (a), S. Pascarelli (b), S. Mobilio (a, c), C. Raghu (d) and D. D. Sarma (d), Phys. Rev. B, 56, 3520 (1997), in press.
(a) INFN, Frascati (Italy)
(b) INFM, Genova (Italy)
(c) Dipartimento di Fisica "E. Amaldi" Universita' di Roma Tre, (Italy)
(d) Solid State and Structural Chemistry Unit, Indian Institute of Science, Bangalore (India)
Large-scale synthesis and structure determination of an organic superconductor
During the last decade the development of new, superconducting materials based on organic molecules has emerged as an important topic for both fundamental as well as application-oriented research. To date, the most important class of organic superconductors are radical cation salts derived from the sulphur heterocycle bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF or ET for short; cf. Figure 61). Technologically relevant characteristics of such superconducting salts are similar to those of industrially used superconducting metal alloys such as NbTi. An example is given in Figure 61.
Recent attempts aimed at the use of organic superconductors or the manufacture of composite materials are undoubtedly a field of great commercial potential. These materials are expected to retain the specific mechanical properties of electrically insulating high-performance plastics, but are electrically conducting at ambient temperature and superconducting at the temperature of liquid helium.
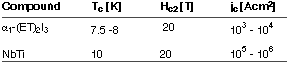
Table 1: Characteristics of an organic superconductor and a superconducting metal alloy.
Tc : superconducting transition temperature;
Hc2: upper critical field; jc: critical current density.
A major obstacle to the successful realisation of the approach outlined above originates from the fact that the needed superconducting material is not readily available and poorly characterised. Compounds of the formal composition (ET)2I3 can exist in more than ten crystallographically distinct phases with properties ranging from semiconducting to metallic to superconducting. An ill-defined, superconducting product, generally referred to as ![]() t-(ET)2I3, was obtained by thermal conversion of the organic metal
t-(ET)2I3, was obtained by thermal conversion of the organic metal ![]() -(ET)2I3, albeit only in mg-quantities. All attempts to determine the structure proved unsuccessful as a consequence of the poor crystal quality obtained after heat treatment.
-(ET)2I3, albeit only in mg-quantities. All attempts to determine the structure proved unsuccessful as a consequence of the poor crystal quality obtained after heat treatment.
Very recently, we succeeded in developing a novel, large-scale preparation method for phase-pure, superconducting ![]() t-(ET)2I3. The process is based on a simple redox-reaction of the organic precursor ET with elemental iodine in organic solvents at elevated temperature.
t-(ET)2I3. The process is based on a simple redox-reaction of the organic precursor ET with elemental iodine in organic solvents at elevated temperature.
The collection of high resolution powder diffraction data at BM16 and subsequent Rietveld refinement allowed the first structure elucidation of this organic superconductor. The average crystal structure and unit cell of ![]() t-(ET)2I3 are presented in Figure 62. The ET molecules are non-planar and arranged in dimers which form loosely-connected stacks along the crystallographic [110] axis. The positively-charged stacks (formal charge: + 0.5 per ET molecule) are separated by layers of negatively charged, linear triiodide (I3-) anions. The carbon atoms at one edge of each ET-molecule show high thermal parameters, indicating disorder of one ethylene group per donor molecule.
t-(ET)2I3 are presented in Figure 62. The ET molecules are non-planar and arranged in dimers which form loosely-connected stacks along the crystallographic [110] axis. The positively-charged stacks (formal charge: + 0.5 per ET molecule) are separated by layers of negatively charged, linear triiodide (I3-) anions. The carbon atoms at one edge of each ET-molecule show high thermal parameters, indicating disorder of one ethylene group per donor molecule.
A typical transition curve as determined by an AC-susceptibility measurement is displayed in Figure 63. The appearance of a diamagnetic signal superconductivity at around 8 K indicates the onset of superconductivity.
The possibility of preparing conducting and even superconducting composite materials, based on a blend of ![]() t-(ET)2I3 (1%) / polycarbonate (99%), could be demonstrated successfully on a laboratory scale. The mechanical properties of the composite and its processability are virtually unchanged with respect to the original polymer.
t-(ET)2I3 (1%) / polycarbonate (99%), could be demonstrated successfully on a laboratory scale. The mechanical properties of the composite and its processability are virtually unchanged with respect to the original polymer.
Apart from the numerous implications for application-oriented research into these technologically promising composite materials, it should also be noted that the easy access to a structurally well-characterised organic superconductor is of considerable importance for fundamental research. To date, neutron scattering and µ-SR measurements, needed for a better understanding of the mechanism of superconductivity in this class of materials, have greatly been hampered by the lack of a convenient source of phase-pure material.
Publication
H. Müller (a), S.O. Svensson (a), A.N. Fitch (a), M. Lorenzen (a) and D.G. Xenikos (a), Adv. Mater., 9, 896 (1997), in press.
(a) ESRF
partners



European Synchrotron Radiation Facility - 71, avenue des Martyrs, CS 40220, 38043 Grenoble Cedex 9, France.